




A osteoartrite é uma doença crônica que danifica a cartilagem e os tecidos circundantes e caracteriza-se por dor, rigidez e perda da função.
A artrite decorrente de lesões na cartilagem das articulações e tecidos circundantes se torna muito comum com o envelhecimento.
Dor, inchaço e supercrescimento ósseo são comuns, bem como a rigidez ao despertar ou após inatividade que desaparece dentro de 30 minutos, especialmente se a articulação for movida.
O diagnóstico se baseia nos sintomas e em radiografias.
O tratamento inclui exercícios e outras medidas físicas, medicamentos que reduzem a dor e melhoram a função e, em casos muito graves, a substituição da articulação ou outra cirurgia.
A osteoartrite, o distúrbio articular mais comum, geralmente começa quando a pessoa tem entre 40 e 50 anos e afeta quase todas as pessoas em algum grau por volta dos 80 anos. Antes dos 40 anos, os homens desenvolvem osteoartrite com mais frequência do que as mulheres, geralmente devido a lesão ou deformidade. Muitas pessoas têm alguma evidência de osteoartrite em radiografias (geralmente aos 40 anos), mas apenas metade dessas pessoas têm sintomas. A partir dos 40 anos e até os 70, as mulheres desenvolvem a doença com mais frequência do que os homens. Depois de 70 anos de idade, a doença se desenvolve em ambos os sexos igualmente.
A osteoartrite é classificada como
Primária
Secundária
Na osteoartrite primária (ou idiopática), a causa não é conhecida (como na grande maioria dos casos). A osteoartrite primária pode afetar apenas certas articulações, como o joelho ou o polegar, ou pode afetar muitas articulações.
Na osteoartrite secundária, a causa é devido a outra doença ou quadro clínico, tal como
Uma infecção
Uma anormalidade articular que surgiu no nascimento
Uma lesão
Um distúrbio metabólico – por exemplo, excesso de ferro no corpo (hemocromatose) ou excesso de cobre no fígado (doença de Wilson)
Uma doença que lesionou a cartilagem articular – por exemplo, artrite reumatoide ou gota
Algumas pessoas que forçam uma articulação ou grupo de articulações, como os trabalhadores de fundição, agricultores, mineiros de carvão e motoristas de ônibus, estão particularmente em risco. O principal fator de risco para osteoartrite do joelho é ter uma profissão que envolve a flexão da articulação. Curiosamente, corrida de longa distância não aumenta o risco de desenvolver a doença. No entanto, se a osteoartrite se desenvolver, esse tipo de exercício, geralmente faz com que o distúrbio piore. A obesidade pode ser um fator importante para o desenvolvimento de osteoartrite, particularmente do joelho e especialmente em mulheres.
Causas da osteoartrite
Normalmente, a cartilagem diminui o nível de atrito nas articulações, protegendo-as do desgaste, mesmo após anos de uso normal, uso excessivo ou lesões. A osteoartrite é causada, na maioria das vezes, por lesão tecidual. Numa tentativa de reparar uma articulação danificada, elementos químicos se acumulam na articulação e aumentam a produção dos componentes da cartilagem, como o colágeno (uma proteína fibrosa resistente no tecido conjuntivo) e proteoglicanos (substâncias que fornecem elasticidade). Em seguida, a cartilagem pode inchar por causa da retenção de líquidos, se tornar macia, começar a degradar e, em seguida, desenvolver fissuras na superfície. Cavidades minúsculas se formam no osso abaixo da cartilagem, enfraquecendo-o.
A tentativa dos tecidos em reparar a lesão pode levar ao crescimento de osso novo e outros tecidos. O osso pode crescer demais nas bordas da articulação, causando colisões (osteófitos) que podem ser vistas e sentidas. Em última análise, a superfície lisa e deslizante da cartilagem torna-se áspera e perfurada, de modo que a articulação já não pode mover-se suavemente e absorver impacto. Todos os componentes da articulação — osso, cápsula articular (tecidos que envolvem a maioria das articulações), tecido sinovial (tecido que reveste a cavidade articular), tendões, ligamentos e cartilagem — apresentam deficiências em várias formas, alterando, assim, a função de articulação.

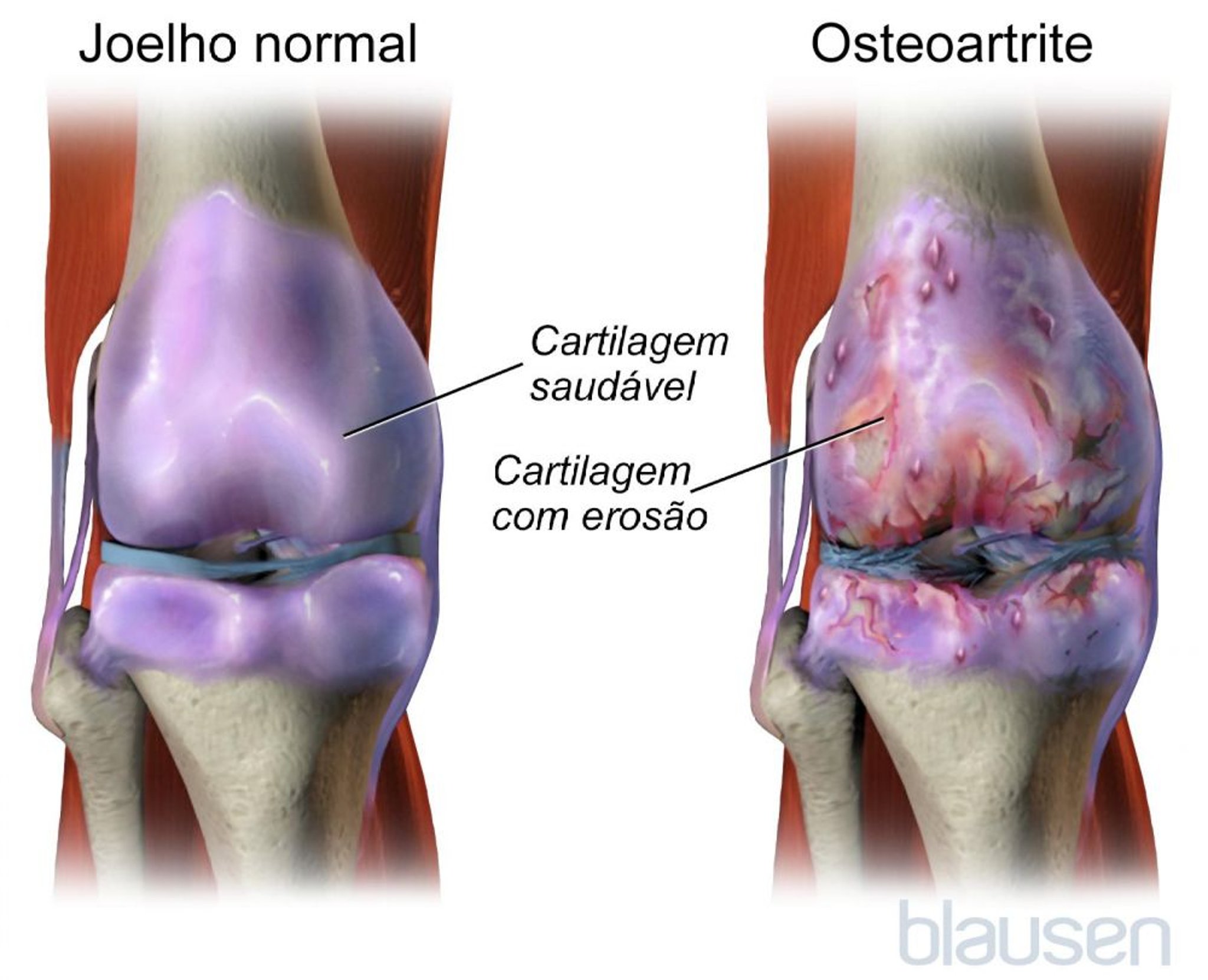
Sintomas de osteoartrite
Geralmente, os sintomas da osteoartrite desenvolvem-se gradualmente e afetam apenas uma ou algumas articulações a princípio. As articulações dos dedos, base dos polegares, pescoço, parte inferior das costas, dedões dos pés, quadris e joelhos são comumente afetadas.
A dor, geralmente descrita como uma dor profunda, é o primeiro sintoma e, quando ocorre nas articulações que suportam peso, geralmente é agravada por atividades que envolvem a sustentação de peso (como ficar em pé). Em algumas pessoas, a articulação pode ficar rígida após o sono ou inatividade, mas a rigidez normalmente desaparece dentro de 30 minutos, especialmente se a articulação for movida.
Conforme a doença provoca mais sintomas, a articulação pode tornar-se menos móvel e não ser capaz de estender-se ou flexionar-se totalmente. O crescimento de osso novo e outros tecidos pode aumentar as articulações. Irregularidades na superfície da cartilagem causam ruídos de raspagem, rangido e crepitação quando elas são movidas e ocorre o desenvolvimento de dolorimento.
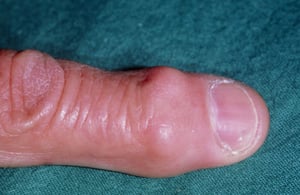
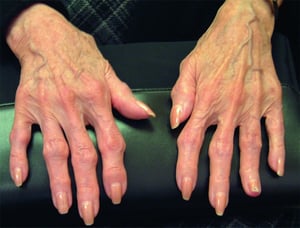
Crescimentos ósseos comumente se desenvolvem nas articulações mais próximas à ponta dos dedos (chamados nódulos de Heberden) ou meio dos dedos (chamados de nódulos de Bouchard).
Em algumas articulações (como o joelho), os ligamentos que circundam e sustentam a articulação ficam estirados e a articulação pode se tornar instável e os músculos que movem a articulação podem ficar enfraquecidos. Alternativamente, o quadril ou joelho podem se tornar rígidos, perdendo sua amplitude de movimento. Movimentar a articulação (especialmente ao ficar em pé, subir escadas ou caminhar) pode ser muito doloroso.
A osteoartrite geralmente afeta a coluna. A dor nas costas é o sintoma mais comum. Normalmente, discos ou articulações lesionados na coluna apenas causam dor e rigidez leves. No entanto, a osteoartrite no pescoço ou na parte inferior das costas pode causar dormência, dor e fraqueza em um braço ou perna se o crescimento excessivo do osso comprimir nervos. O crescimento excessivo de osso pode estar dentro do canal medular na região lombar (estenose lombar da coluna vertebral), pressionando os nervos antes de eles saírem do canal em direção às pernas. Essa pressão pode causar dor nas pernas depois de uma caminhada, sugerindo incorretamente que a pessoa tem uma diminuição do suprimento sanguíneo para as pernas (claudicação intermitente). Raramente, os crescimentos ósseos no pescoço comprimem o esôfago, dificultando a deglutição.
A osteoartrite pode ser estável por muitos anos ou pode progredir muito rapidamente, mas, na maioria das vezes, progride lentamente depois que os sintomas se desenvolvem. Muitas pessoas desenvolvem algum grau de deficiência.
Pessoas que desenvolvem uma articulação vermelha, quente e inchada devem ser avaliadas por um médico, uma vez que esses episódios podem não ser causados pela osteoartrite e podem resultar de uma infecção ou gota.
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
© Springer Science+Business Media


DR P. MARAZZI/SCIENCE PHOTO LIBRARY


© Springer Science+Business Media
Diagnóstico de osteoartrite
Radiografias
O médico faz o diagnóstico de osteoartrite baseado nos sintomas característicos, no exame físico e na aparência das articulações em radiografias (como crescimento ósseo e estreitamento do espaço articular). No entanto, as radiografias não são muito úteis para a detecção precoce da osteoartrite porque elas não mostram alterações na cartilagem, local onde ocorrem as primeiras anormalidades. Além disso, alterações na radiografia geralmente não correspondem estritamente aos sintomas da pessoa. Por exemplo, uma radiografia pode mostrar apenas uma pequena alteração em uma pessoa que tem sintomas graves, ou pode mostrar inúmeras alterações em uma pessoa que tem poucos ou nenhum sintoma.
A ressonância magnética (RM) pode revelar alterações precoces na cartilagem, mas raramente é necessária para o diagnóstico.
Não há exames de sangue para o diagnóstico de osteoartrite, embora certos exames de sangue possam ajudar a descartar outras doenças.
Se uma articulação estiver inchada, os médicos podem injetar um anestésico para insensibilizar a área e inserir uma agulha na articulação para coletar uma amostra do líquido articular. O líquido é examinado para diferenciar osteoartrite de outras doenças articulares comuns, como infecção e gota.
Tratamento de osteoartrite
Medidas físicas, incluindo fisioterapia e terapia ocupacional, proteção articular e perda de peso apropriada
Medicamentos
Cirurgia
Os principais objetivos do tratamento da osteoartrite são
Aliviar a dor
Manter a flexibilidade da articulação
Otimizar a função articular e geral
Esses objetivos são alcançados principalmente através de medidas físicas que envolvem exercícios de força, flexibilidade, resistência e reabilitação (fisioterapia e terapia ocupacional). As pessoas aprendem como modificar suas atividades diárias pode ajudá-las a viver com osteoartrite. O tratamento adicional inclui medicamentos, cirurgias (para algumas pessoas) e novas terapias.
Medidas físicas
Exercícios adequados, incluindo exercícios de alongamento, fortalecimento e posturais, ajudam a manter a cartilagem saudável, aumentar a amplitude de movimento de uma articulação e, mais importante, fortalecer os músculos ao redor das articulações, para que possam absorver melhor o esforço. O exercício pode, às vezes, retardar o agravamento da osteoartrite do quadril e do joelho. Os médicos costumam recomendar que as pessoas se exercitem na água (como em uma piscina), pois a água poupa as articulações do esforço.
Exercícios de alongamento devem ser feitos diariamente.
O exercício deve ser equilibrado com alguns minutos de descanso em caso de articulações dolorosas (a cada quatro a seis horas durante o dia), mas a imobilização de uma articulação tende mais a agravar a doença do que aliviá-la.
Usar cadeiras, poltronas, colchões e assentos de carro excessivamente macios pode piorar os sintomas.
As pessoas não devem colocar travesseiros sob os joelhos quando deitadas porque isto pode fazer com que os músculos do quadril e joelhos fiquem rígidos. (Essa recomendação contrasta com a recomendação de que as pessoas com dor lombar e ciática devem colocar um travesseiro entre os joelhos. Para tais pessoas, utilizar um travesseiro alivia o stress na região lombar e no quadril [consulte Ciática].)
Geralmente se recomenda usar os assentos do carro erguidos, usar cadeiras de encosto reto com assento relativamente alto (como cadeiras da cozinha ou sala de jantar), colchões firmes e placas na cama (disponíveis em muitas madeireiras), bem como usar sapatos com suportes ortóticos ou calçados esportivos.
Um elevador de assento sanitário pode ajudar as pessoas que têm osteoartrose dolorosa dos joelhos ou quadris a se levantarem mais facilmente e com menos desconforto, principalmente se seus músculos são fracos.
Para osteoartrite da coluna, exercícios específicos às vezes ajudam e podem ser necessários coletes ortopédicos para as costas quando a dor é grave. Os exercícios devem incluir tanto o fortalecimento muscular como exercícios aeróbicos de baixo impacto (como fazer caminhadas, natação, usar o aparelho elíptico e bicicleta estacionária). Se possível, as pessoas devem manter as atividades diárias normais e continuar a exercer suas atividades normalmente, como divertir-se e trabalhar. No entanto, pode ser necessário ajustar as atividades físicas para limitar o levantamento e a flexão de maneiras que possam agravar a dor da osteoartrite.
Outras medidas adicionais podem ajudar a aliviar a dor e ajudar as pessoas a viver com osteoartrite. Incluem
Fisioterapia, frequentemente com terapia com calor, como compressas quentes, e terapia ocupacional podem ser úteis.
Exercícios para amplitude de movimento feitos suavemente em água morna são úteis, pois o calor melhora a função muscular, reduzindo a rigidez e o espasmo muscular.
Terapia com calor, como compressas quentes ou uma toalha úmida e quente, pode ser aplicada nas articulações afetadas. (Para evitar queimaduras, as pessoas devem evitar uma compressa elétrica muito quente ou deixar ela no lugar por muito tempo.)
Frio também pode ser aplicado para reduzir a dor causada pela piora temporária em uma articulação.
Palmilhas (órteses), sapatos de apoio ou calçados esportivos podem ajudar a reduzir a dor causada ao andar.
Equipamentos especiais (por exemplo, bengalas, muletas, andadores, colar cervical ou tensor elástico nos joelhos para proteger as articulações ou um assento fixo colocado em uma banheira para evitar muito alongamento durante o banho) devem ser usados conforme necessários.
Perda de peso pode aliviar alguma pressão sobre as articulações e melhorar a mecânica.
Estimulação elétrica, como a estimulação elétrica transcutânea do nervo (transcutaneous electrical nerve stimulation, TENS), pode ajudar a aliviar a dor.
Acupuntura libera vários mensageiros químicos no cérebro (neurotransmissores), que servem de atenuadores naturais da dor e podem ser úteis.
Massagem por terapeutas treinados e tratamento térmico profundo com diatermia ou ultrassonografia podem ser úteis.
Medicamentos
São usados medicamentos para complementar exercícios e fisioterapia. Os medicamentos, que podem ser utilizados em combinação ou isoladamente, não alteram diretamente o curso da osteoartrite. Eles são usados para reduzir os sintomas e, assim, permitir que as atividades diárias mais normais sejam realizadas.
Um medicamento para dor simples (analgésico), como o paracetamol, usado antes de atividades que causam desconforto ou usado regularmente para aliviar o desconforto articular mais constante, pode ser tudo o que é necessário para dor leve a moderada. Embora os efeitos colaterais não sejam comuns, as pessoas não devem tomar paracetamol em doses mais elevadas do que as recomendadas, particularmente se tiverem uma doença hepática ou consumirem uma quantidade considerável de álcool. Ao tomar paracetamol, as pessoas também devem certificar-se de não tomar simultaneamente um dos numerosos medicamentos sem prescrição médica que contêm acetaminofeno.
Às vezes, porém, as pessoas podem precisar de um analgésico mais potente, como o tramadol.
Alternativamente, um anti-inflamatório não esteroide (AINE) pode ser tomado para diminuir a dor e o inchaço. AINEs reduzem a dor e a inflamação nas articulações e podem ser usados em combinação com outros analgésicos. AINEs também vêm em formas de gel e creme que podem ser espalhados na pele (como o diclofenaco em gel a 1%) sobre as articulações das mãos e joelhos e podem ajudar a aliviar os sintomas. No entanto, os AINEs têm um maior risco de efeitos colaterais graves do que o paracetamol quando usados em longo prazo. Pessoas que tomam AINEs por via oral frequentemente também tomam um medicamento para proteger o revestimento gástrico e também podem ter sua função renal e pressão arterial monitoradas.
Às vezes, podem ser necessários outros tipos de medicamentos para dor. Por exemplo, um creme feito a partir da pimenta caiena – seu ingrediente ativo é a capsaicina – pode ser aplicado diretamente na pele sobre a articulação. Os médicos também podem recomendar compressas de lidocaína para alívio da dor, mas não existem evidências de que essas compressas sejam eficazes. Duloxetina, um tipo de antidepressivo tomado por via oral, pode reduzir a dor causada por osteoartrite.
Relaxantes musculares (geralmente em doses baixas) ocasionalmente aliviam a dor causada por músculos tensos por suportarem as articulações afetadas pela osteoartrite. Em pessoas idosas, no entanto, eles podem causar mais efeitos colaterais do que alívio.
Se uma articulação repentinamente se tornar inflamada, inchada e dolorosa, a maior parte do líquido no interior da articulação pode ter de ser removido e uma forma especial de cortisona pode ser injetada diretamente na articulação. Esse tratamento pode proporcionar alívio temporário da dor e aumento da flexibilidade das articulações em algumas pessoas.
Uma série de uma a cinco injeções semanais de hialuronato (uma substância semelhante ao líquido articular normal) na articulação do joelho pode fornecer algum alívio da dor em algumas pessoas por períodos prolongados de tempo. Essas injeções não devem ser administradas com mais frequência do que a cada seis meses. Injeções de hialuronato são menos eficazes em pessoas com osteoartrite grave e não retardam a progressão da artrite.
Vários suplementos nutricionais (como sulfato de glucosamina e sulfato de condroitina) têm sido testados quanto ao benefício potencial no tratamento de osteoartrite. Até agora, os resultados são confusos, o benefício potencial do sulfato de glucosamina e do sulfato de condroitina no tratamento da dor é incerto e não parecem ter alterado a progressão da lesão articular. Não há boa evidência de que quaisquer outros suplementos nutricionais funcionem.
Cirurgia
O tratamento cirúrgico pode ajudar quando outros tratamentos falham em reduzir a dor ou em melhorar a função. Algumas articulações, mais comumente o quadril e o joelho, podem ser substituídas por uma articulação artificial. A substituição, particularmente a do quadril, é geralmente muito bem-sucedida, quase sempre melhora o movimento e a função e reduz drasticamente a dor. Portanto, a substituição da articulação deve ser considerada quando a dor é incontrolável e a função torna-se limitada. Como uma articulação artificial não dura para sempre, o procedimento é geralmente adiado em pessoas muito jovens para que a necessidade de substituições repetidas seja minimizada. Se os outros tratamentos forem ineficazes, podem ser feitos procedimentos cirúrgicos para ajudar a aliviar os sintomas da osteoartrite de coluna e pescoço, particularmente a compressão de nervo. Os benefícios de procedimentos cirúrgicos artroscópicos limitados para osteoartrite do joelho, como o reparo do menisco ou a reconstrução dos ligamentos do joelho, são incertos.
Uma variedade de métodos que restauram as células dentro da cartilagem tem sido utilizada em pessoas mais jovens com osteoartrite (geralmente causada por uma lesão) para ajudar a curar pequenos defeitos na cartilagem. No entanto, estes métodos ainda demonstraram ser valiosos quando os defeitos da cartilagem são extensos, como normalmente ocorre em pessoas idosas.
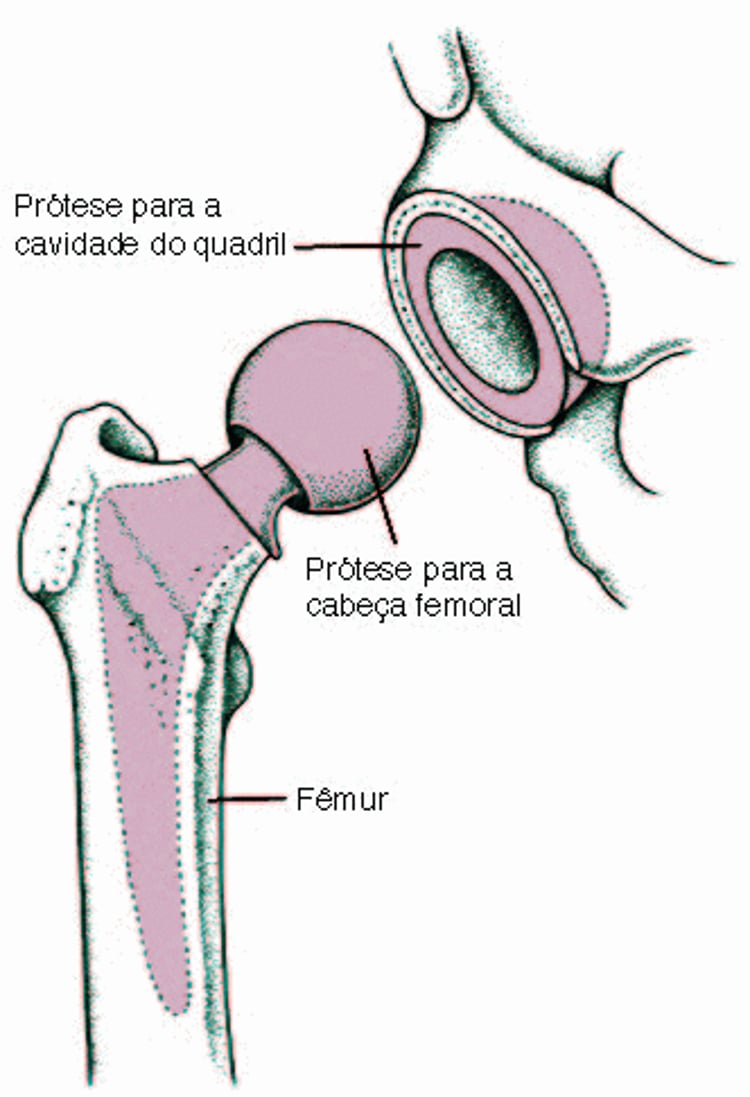
Substituição de todo o quadril (substituição total de quadril)
Por vezes, é preciso substituir toda a articulação do quadril. A articulação completa do quadril é a extremidade superior (cabeça) do osso da coxa (fêmur) e a superfície de encaixe onde a cabeça do osso da coxa se encaixa. Esse procedimento é chamado substituição total do quadril ou artroplastia total do quadril. A cabeça do osso da coxa é substituída por uma peça em forma de esfera (prótese), feita em metal. A prótese possui uma estrutura central forte que se encaixa dentro do centro do osso da coxa. O encaixe é substituído por um invólucro de metal revestido de plástico durável. |

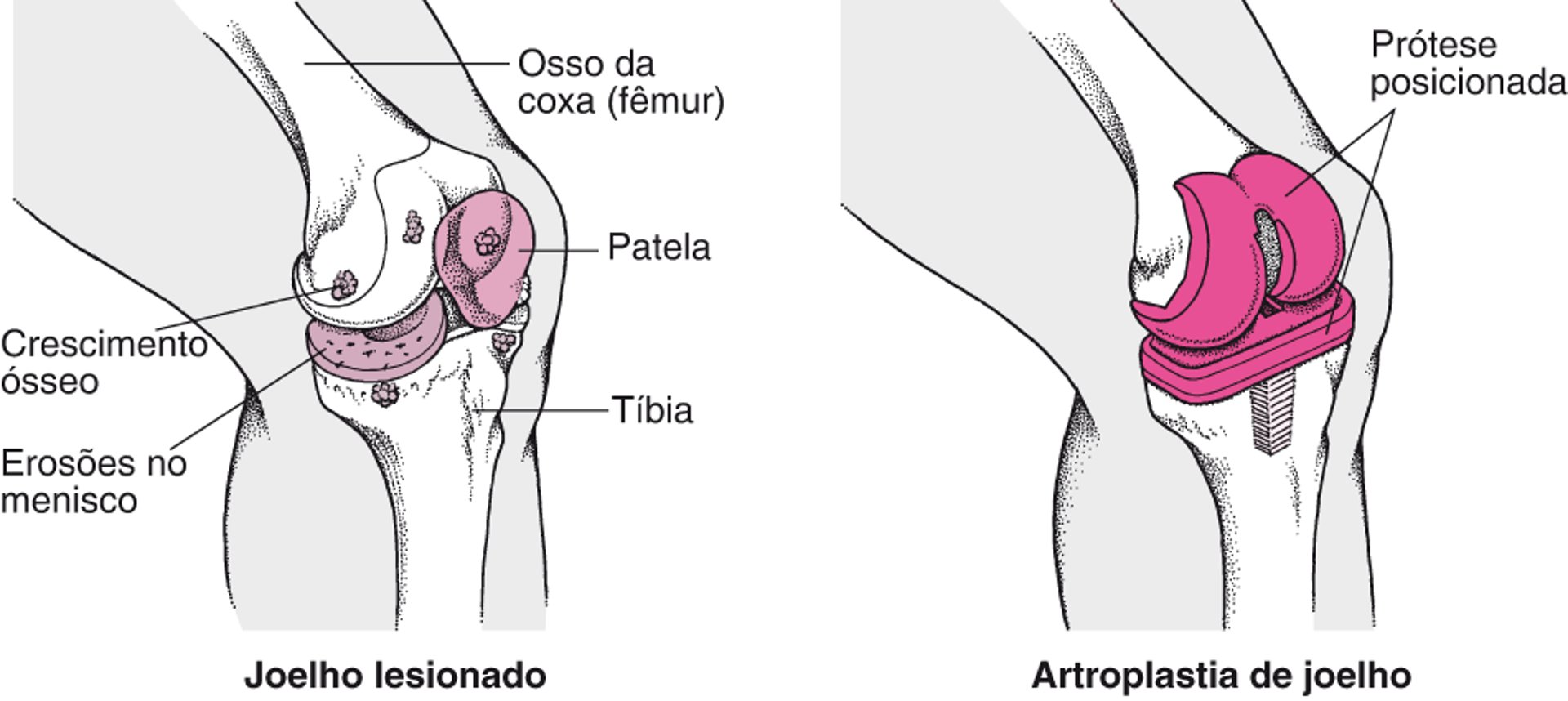
Substituição de joelho
A articulação do joelho danificada pela osteoartrite pode ser substituída por uma articulação artificial. Após uma anestesia geral ser administrada, o cirurgião faz uma incisão sobre o joelho danificado. A rótula (patela) pode ser removida e as extremidades do osso da coxa (fêmur) e canela (tíbia) são polidas de modo que as partes da articulação artificial (prótese) possam ser fixadas mais facilmente. Uma parte da articulação artificial é inserida no osso da coxa, a outra parte é inserida na tíbia, e, em seguida, as peças são cimentadas no local do implante. |

Destaque para Idosos: Osteoartrite
Muitos mitos sobre osteoartrite permanecem. Por exemplo, há o mito de ela ser parte inevitável do envelhecimento, como os cabelos grisalhos e as modificações na pele, e resultar em pouca deficiência e o mito de o tratamento não ser eficaz. A osteoartrite se torna mais comum com o envelhecimento. Por exemplo, conforme a idade, ocorre o seguinte:
No entanto, a osteoartrite não é uma parte inevitável do envelhecimento. Ela não é causada simplesmente pelo desgaste que ocorre com os anos de utilização da articulação. Outros fatores podem incluir lesão única ou repetida, movimento anormal, distúrbios metabólicos, infecção articular ou outro distúrbio da articulação. Está disponível tratamento eficaz, como medicamentos para dor (analgésicos), exercícios, fisioterapia e, em alguns casos, cirurgia. Lesão nos ligamentos também é comum com o envelhecimento. Os ligamentos, que se ligam as articulações, tendem a tornar-se menos elásticos com a idade, fazendo as pessoas sentirem suas articulações apertadas ou rígidas. Essa mudança resulta de alterações químicas nas proteínas que compõem os ligamentos. Consequentemente, a maioria das pessoas se tornam menos flexíveis à medida que envelhecem. Os ligamentos tendem a se romper mais facilmente, e quando eles se rompem, se curam mais vagarosamente. As pessoas idosas devem ter o seu regime de exercício avaliado por um treinador ou médico, de modo que os exercícios que podem romper ligamentos sejam evitados. Às vezes, a dor causada pela osteoartrite não pode ser aliviada por um analgésico simples, como o paracetamol. Podem ser necessários analgésicos mais potentes, como tramadol, mas os médicos receitam esses medicamentos apenas quando necessário, para evitar problemas com efeitos colaterais e possível dependência. No entanto, esses medicamentos podem causar confusão em pessoas idosas. Medicamentos anti-inflamatórios não esteroides (AINEs), que são espalhados na pele sobre a articulação afetada, podem ser uma melhor opção para as pessoas idosas. Uma menor quantidade de AINEs é absorvida do que se for tomado por via oral, minimizando o risco de efeitos colaterais. |
Mais informações
Os seguintes recursos em inglês podem ser úteis. Vale ressaltar que O MANUAL não é responsável pelo conteúdo deste recurso.
Arthritis Foundation: Informações sobre osteoartrite e outros tipos de artrite e tratamentos disponíveis, dicas de estilo de vida e outros recursos




